Метаболизмот на јаглени хидрати кај доенчиња

Cодржина
- Видео: Повреда на метаболизмот на јаглени хидрати со деца
- болести на гликогенските депоа
- Видео: Филмот на манастирот. Простите јаглехидрати. Висок гликемиски индекс
- Нарушувања на метаболизмот на фруктоза
- Нарушувања на пируват метаболизам
- Видео: Патофизиологија на метаболизмот на јаглени хидрати (предавање). t.n. alhendi. дел 2/2
- Видео: Вродени грешки на метаболизмот на јаглени хидрати. Клинички карактеристики, дијагноза, третман
- Други нарушувања на метаболизмот на јаглени хидрати
Галактоземија - автосомно рецесивно нарушување се јавува во 1 дете до 60 000 живородени деца.
Во класичната, почеста форма на недостаток на галактоза 1-fosfaturidiltransferazy што води до таложење на галактоза, галактоза 1-фосфат и galaktiola ткиво. Помалку често се предизвикани од недостаток на галактокиназа галактоземија или епимераза a. За прием на лактоза се појават кај новородени храна повраќање, дијареа, хипербилирубинемија, хепатоспленомегалија, ренална тубуларна дисфункција, хепатална инсуфициенција, катаракта. Галактоза во крвта на болните деца.
Дијагнозата се потврдува со мерење на активноста на галактоза 1-fosfatgransferazy во црвените крвни клетки кои ќе биде вистина ако тоа не е проследен и со црвени крвни клетки трансфузија.
Морфолошки промени вклучуваат изговара со прогресивна стеатоза на црниот дроб psevdoatsinarnoy преуредување структура резени пролиферација на жолчните канали, холестаза, фокална некроза со исходот во цироза.
Исто така, постојат островските клетки хиперплазија, вакуолација nephrothelial неспецифични исхемично оштетување на мозокот во форма на невронот смрт, глиоза, едем.
Морфолошки промени на хередитарна фруктоза интолеранција и Тирозинемија се слични (види. Подолу).
Повреди на метаболизмот на фруктоза. Претставуваат болест со автосомно рецесивен начин на наследување, се манифестира кај новороденото рок за прием со храна. Манифестира со повраќање, гадење, хепатомегалија, хепатална инсуфициенција, хипогликемија, млечна ацидоза. Стеатоза, холестаза, портална фиброза и пролиферацијата cholangioles со трансформација во цироза.
Glycogenoses - група на наследни нарушувања на метаболизмот на јаглени хидрати кои произлегуваат од мутации во неколку гени кои кодираат ензими кои се регулира синтезата и разградувањето на гликоген во едноставни шеќери и нормално или абнормална акумулација на гликоген во клетките на многу ткива. На класификација се базира на дефект на еден специфичен ензим. Наследува автосомно рецесивно, освен гликогеноза тип VIII, секс-поврзани.
На различни форми на болеста на депонирање на гликоген (тип 14) се одразува на учество во гликоген метаболизмот на голем број на гени.
Поголемиот дел од пациентите имаат системски манифестации забележани во некои форми на неуспехот на одделни органи. Карактеризира со симптоми на миопатија, како и епизоди на хипогликемија, кардиомегалија развој.
Видео: Повреда на метаболизмот на јаглени хидрати со деца
болести на гликогенските депоа
болести на гликогенските депоа (GSDs) поради недостаток на ензими кои се вклучени во синтезата или уништување glikogena- недостатоци може да се случи во црниот дроб, болки во мускулите и да предизвика хипогликемија или таложење на абнормални количини или видови на абнормален гликоген во ткивата.
Видео: Филмот на манастирот. Простите јаглехидрати. Висок гликемиски индекс
Наследство во GSDs автосомно рецесивно освен видови ДДЦК VIII / IX се во X-поврзани. Инциденцата се проценува на околу 1/25 000 деца, а тоа може да се потцени, бидејќи светлината субклинички форми не секогаш се дијагностицира.
Возраст на почетокот, клинички манифестации и тежината зависи од видот.
Дијагнозата се потврдува со значително намалување на ензимската активност во црниот дроб (тип I, III, VI и VIII / IX), мускулите (тип lib, III, VII и VIII / IX), кожата фибробласти (Видови ХА и IV) или еритроцити (Тип VII) отсуство или се зголеми активноста на венски лактат / исхемија подлактицата (тип V и VII). Прогноза и третман се разликуваат во зависност од видот, но третманот често вклучува и додатоци во исхраната со пченкарен скроб да се обезбеди постојан извор на гликоза во црниот дроб форми на ДДЦК, и вежбање за да се спречи мускулни форми.
Дефекти во гликолизата (ретко) може да предизвика синдром сличен на GSDs. Фосфоглицерат дефицит на киназа, фосфоглицерат мутаза и лактат миопатија мимик Вид на ДДЦК V и VII- дефицити на гликоза транспортен протеин 2 (јаболковина Фанкони - Bickel) мимик ДДЦК хепатопатија други видови (на пример, 1.111, IV, VI).
Нарушувања на метаболизмот на фруктоза
Недостаток метаболизира ензимот фруктоза, може да биде асимптоматски или да предизвика хипогликемија.
Фруктоза е моносахаридот којшто е присутен во високи концентрации во овошје и мед и формирање на дел од сахароза и сорбитол.
Недостаток на фруктоза-1-фосфат алдолаза (алдолаза Б). Овој недостаток е клинички синдром, хередитарна фруктоза нетолеранција. Наследувањето е автозомно retsessivnoe- инциденцата се проценува на 1/20 000 деца. Бебињата останат здрави, а не консумираат фруктоза. Долготрајната употреба може да предизвика цироза, ментално нарушување и ренална тубуларна ацидоза проксимално на отсуство на фосфати во урина и гликоза.
Дијагноза врз основа на симптомите сугерираат во поглед на неодамнешната доза на фруктоза и потврди ензим анализа на ткиво на црниот дроб биопсија или со индуцирање на хипогликемија интравенска инфузија на фруктоза во количина од 200 mg / kg.
Краткорочни третман е да се добие на гликоза gipoglikemii- долгорочен третман е отстранување на потрошувачката на фруктоза, сахароза и сорбитол. Многу пациенти развој на природен фруктоза интолеранција на храна. Во третманот на прогнозата е одлична.
фруктокиназа дефицит. Овој недостаток предизвикува бенигно зголемување на крв и урина ниво на фруктоза (бенигна фруктозурија). Наследувањето е автозомно retsessivnoe- инциденца е околу 1/130 000 деца.
Статус асимптоматски и дијагностициран со можност за откривање на уринарниот neglyukozovosstanavlivayuschego супстанции.
Недостаток на фруктоза-1, 6-бифосфатаза. Овој недостаток компромиси на глуконеогенезата и доведува до постот хипогликемија, кетоза и ацидоза. Недостаток може да биде фатална за бебиња. Наследство автосомно retsessivnoe- фреквенција е непозната. Постојат епизоди на треска.
Прва Третманот се состои од орална или интравенска администрација на глукоза. Толеранција на глад обично се зголемува со возраста.
Нарушувања на пируват метаболизам
Неможноста да се метаболизираат пируват води кон развој на лактатна ацидоза и разни абнормалности на централниот нервен систем.
Видео: Патофизиологија на метаболизмот на јаглени хидрати (предавање). T.N. Alhendi. дел 2/2
Пируват е важна супстрат на метаболизмот на јаглени хидрати.
Недостаток на пируват дехидрогеназа. Piru vatdegidrogenaza е multienzyme комплекс одговорен за формирање на ацетил-CoA од пируват за Кребсовиот циклус. Недостаток доведува до зголемување на нивото на пируват, а со тоа зголемување на нивото на млечна киселина. X-поврзано наследување или авто-хромозомски рецесивно.
Видео: Вродени грешки на метаболизмот на јаглени хидрати. Клинички карактеристики, дијагноза, третман
Клиничките манифестации се разликуваат во тежината, но вклучуваат лактична ацидоза.
Дијагнозата се потврдува со ензимска анализа активност на кожата фибробласти ДНК анализа овие два методи.
Јасно е дека не постои ефикасен третман, додека на диета со малку јагленохидрати или ketogenic исхрана и додатоци во исхраната тиамин се корисни за некои пациенти.
Недостаток на пируват карбоксилазата. Недостаток може да биде примарна или секундарна на недостаток golokarboksilazy синтетаза биотин или biotinidazy- автосомно рецесивно наследување на, и двете варијанти доведе до лактична ацидоза.
Инциденцата на примарниот дефицит <1/250 000 родов, но может быть выше у некоторых американских индейских народов. Задержка психомоторного развития с эпилептическими припадками и спастичность являются основными клиническими проявлениями. Нарушения лабораторных показателей включают гипераммониемию- молочный ацидоз, кетоацидоз.
Клиничките манифестации на секундарниот дефицит се слични и се неуспехот да се напредува, напади и други органски ацидурија.
Ефективен третман не постои, но некои пациенти со примарен дефицит и сите лица кои имаат просек тежината на болеста, тоа е потребно да се додаде биотин 5-20 mg орално еднаш дневно.
Други нарушувања на метаболизмот на јаглени хидрати
Недостаток на фосфоенолпируват carboxykinase (261.680) дава глуконеогенезата и да доведе до симптоми и знаци слични на хепатална форма гликогеноза, но без акумулација на гликоген во црниот дроб.
Други дефицити се неуспехот гликолитични ензими или ензими од пентозо фосфат патека. Типични примери се недостаток на пируват киназа и гликоза-6-фосфат дехидрогеназа (G6PD), која може да доведе до развој на хемолитична анемија. Верник синдром - Корсаков предизвикани делумно неуспехот transketolase пентозо фосфат патека е ензим кој се бара тиамин и како кофактор.
 Laktofiltrum дијареа
Laktofiltrum дијареа Галактоземија
Галактоземија Гликоген за складирање болест мали сипаници, Андерсен McArdl. болест Hersey е, Томсон, контејнери
Гликоген за складирање болест мали сипаници, Андерсен McArdl. болест Hersey е, Томсон, контејнери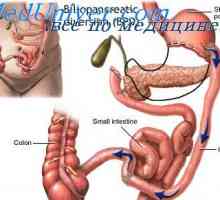 Варење на јаглени хидрати. Низа варење на јаглени хидрати во гастроинтестиналниот тракт
Варење на јаглени хидрати. Низа варење на јаглени хидрати во гастроинтестиналниот тракт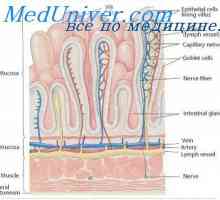 Јаглени хидрати апсорпција во цревата. На апсорпција на протеините во стомакот
Јаглени хидрати апсорпција во цревата. На апсорпција на протеините во стомакот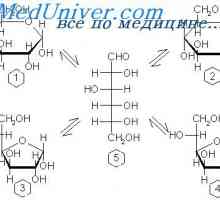 Физиологија на метаболизмот на гликозата. Транспорт на гликоза низ клеточната мембрана
Физиологија на метаболизмот на гликозата. Транспорт на гликоза низ клеточната мембрана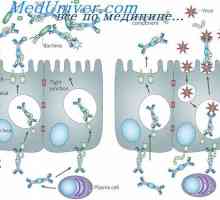 На секреција на имуноглобулини. Фази секреција антитело
На секреција на имуноглобулини. Фази секреција антитело Автосомно рецесивно полицистична бубрежна болест кај децата. Дијагноза и терапија
Автосомно рецесивно полицистична бубрежна болест кај децата. Дијагноза и терапија Неонатална жолтица со метаболички нарушувања
Неонатална жолтица со метаболички нарушувања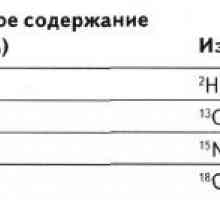 Методи за оценување на метаболизмот на јаглени хидрати и масти во телото
Методи за оценување на метаболизмот на јаглени хидрати и масти во телото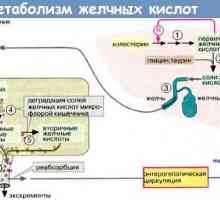 Прогресивна фамилијарна интрахепатична холестаза (PFIC) видови, дијагностика
Прогресивна фамилијарна интрахепатична холестаза (PFIC) видови, дијагностика Лактулозата (lastulosum). Лактулозата се однесува на синтетички дисахарид. Кога се дава орално не…
Лактулозата (lastulosum). Лактулозата се однесува на синтетички дисахарид. Кога се дава орално не…- Ehovist (echovist). Ehovist-200 е суспензија на микронизиран d-galaktoy. Кога е суспендиран…
- Здравствени енциклопедија, болест, лекови, лекар, аптека, инфекција, извадоци, секс, гинекологија,…
- Терапија, болести на дигестивниот систем
 Метаболички нарушувања на циклусот урична киселина
Метаболички нарушувања на циклусот урична киселина Конгенитални хепатална фиброза
Конгенитални хепатална фиброза Размена прекршувања на метали
Размена прекршувања на метали Исхрана и наследноста дете
Исхрана и наследноста дете Метаболички нарушувања на метаболизмот на урична киселина и метали
Метаболички нарушувања на метаболизмот на урична киселина и метали Цироза на црниот дроб кај децата симптоми
Цироза на црниот дроб кај децата симптоми
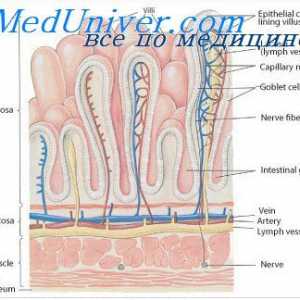 Јаглени хидрати апсорпција во цревата. На апсорпција на протеините во стомакот
Јаглени хидрати апсорпција во цревата. На апсорпција на протеините во стомакот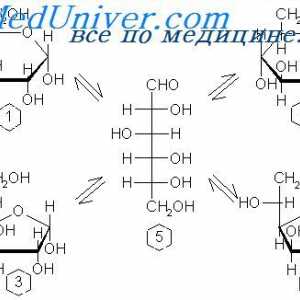 Физиологија на метаболизмот на гликозата. Транспорт на гликоза низ клеточната мембрана
Физиологија на метаболизмот на гликозата. Транспорт на гликоза низ клеточната мембрана Автосомно рецесивно полицистична бубрежна болест кај децата. Дијагноза и терапија
Автосомно рецесивно полицистична бубрежна болест кај децата. Дијагноза и терапија Неонатална жолтица со метаболички нарушувања
Неонатална жолтица со метаболички нарушувања Галактоземија
Галактоземија Размена прекршувања на метали
Размена прекршувања на метали Исхрана и наследноста дете
Исхрана и наследноста дете