Женски псевдохермафродитизам

Cодржина
Видео: 32-годишен маж се смета себеси за хермафродит - Јас soromlyus Своге tіla - 05/15/15
Таквите пациенти имаат нормален јајниците и деривати Mullerian комбинација со неизвесна надворешните гениталии.
Во отсуство на тестисите микроб masculinizing женски под влијание на покачени концентрации на андрогени во крвта на мајката. Степенот на вирилизација зависи од степенот на развојот на фетусот во времето на изложеност на вишокот на андрогените. По 12 недели од бременоста ефекти на андрогените предизвикува само хипертрофија на клиторисот. Во ретки случаи, двојна надворешните гениталии како формира под влијание на андрогените, се резултат на влијанието на други тератогени.
Конгенитална адренална хиперплазија
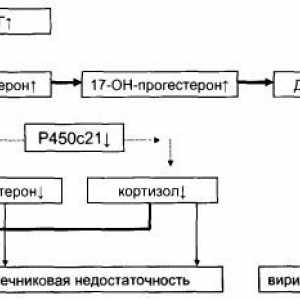
Причината за повеќето случаи на женски псевдохермафродитизам и околу 50% од случаите на двојна гениталии е конгенитална адренална хиперплазија. Постојат 6 главни видови на конгенитална адренална хиперплазија, сите тие се автосомно рецесивно болести. Заедничка карактеристика на сите шест видови е синтеза на кортизол дефект, што резултираше со зголемена содржина на ACTH и, според тоа, има адренална хиперплазија. Болеста се јавува и кај мажите и жените, но мажите ретко се дијагностицира при раѓање, освен ако тие не се појавиш двојна genitalii- болест е solteryayuschim и се манифестира адренална криза, снимено или скрининг на населението за сите новороденчиња, или со скрининг на високо ризичните групи - браќа и сестри на лица со оваа болест. 21 неуспех предизвикува вирилизација-хидро-ksilazy и надбубрежните 11-хидроксилаза.
Неодамна, во својата студија на Милер и сор. што е опишано олицетворение автозомно рецесивно конгенитална адренална хиперплазија произлегуваат како резултат на мутација на генот кој го кодира синтеза на Р450 оксидоредуктаза, Flavio еден протеин, кој е електрони донатор на сите микрозомални ензими P450 tsitrohroma, вклучувајќи ги и 17-хидроксилаза / 17,20-лиаза, 21-хидро- ksilazu и ароматаза. Мајка на засегнатите фетуси се изложени на ризик од вирилизација време на бременоста. 46, XX пациенти со недостатокот на надбубрежните жлезди и јајниците стероидогенеза имаат двојно гениталии, додека пациентите со кариотип 46, XY имаат нецелосни maskulyanizatsiyu надворешните гениталии. Некои пациенти имаат синдром скелетни дисморфија (синдром Antle-Bixler). Пациенти со мутации P450 оксидоредуктаза (на соба) се изложени на ризик за развој на адренална инсуфициенција, особено за време на фебрилни болести со треска или за време на операцијата. На ниски позадина или нормална содржина на C19 стероиди опише покачена серумска концентрација на прогестерон и 17-OH прогестерон во одговор на зголемување на производството на АЦТХ. Сепак P450 оксидоредуктаза неуспехот дијагноза во отсуство на болест на коските врз основа на анализа на содржината на стероиди во урината по маса спектроскопија, проследено со потврда за мутација на ДНК. Општо земено, при анализата на содржината на стероиди во урината откри релативно низок износ на метаболитите на андрогените и зголемена екскреција на метаболитите на прогестерон и прегненолон.
Дефектот-хидроксистероид дехидрогеназа тип II, 17-хидроксилаза (17,20-лиаза), steroidogenic акутен регулаторен протеин (ѕвезда) и P450scc (страничен ланец пресекување) се појавува и блокада на кортизол синтеза на сексуални стероиди во адреналната и гонадите. Овие видови се појавуваат најчесто некомплетна маскулинизација на момчиња и малку или воопшто не вирилизација кај девојчињата. Затоа, овие форми ќе се дискутира, првенствено како машки псевдохермафродитизам.
Хидроксилаза дефицит R450s21
21-хидроксилаза активност е регулирана R450s21, микрозомални цитохром P450. Недостаток на овој ензим предизвикува најчест тип на надбубрежните кортекс, со вкупна стапка на 01:14 000 живородени жители на Европската раса. Со 21-хидроксилаза дефицит кај повеќе од 90% од пациентите со VDKN во Северна Америка и Европа. кодирање на генот за синтеза на 21-хидроксилаза, се наоѓа на краткиот крак на хромозомот 6, во близина на генот C4 (комплемент) помеѓу HLA-B и HLA-D. Кога ДНК анализа во овој локус, се идентификувани два гена, определени како R450s21A (CYP21P) и />450s21V (CYP21), кои се во тандем два гени за да го дополни Ц4А и C4b, и два гени Tb Ха и, надредениот една врз друга на спротивните R450s21A R450s21V делови и ДНК. Гени кои кодираат? А и Xb ekstatsellyulyarny матрикс протеини тенасцин-X.
R450s21A е нефункционален "pseudogene" (vol. Е. Тоа нема шематски структурни низа, и тоа не се кодираат функционирање на 21-хидроксилаза). 75% од пациентите со класичен неуспех R450s21 имаат точкести мутации што резултира со мали промени во дел R450s21V хомологни база секвенца R450s21A нефункционални гени - затоа постои "microgeny конверзија". Околу 15% од изразените гени 21- OH неуспехот забележано бришење дел помеѓу 3-от и 8-ми егзон pseudogene R450s21 А делот хомологни на генот 21-HB, што резултира со не-функционални гени на соединението 21-IT / 21-CAC. Ова доведува до неусогласеност и нееднаквиот премин. Во остатокот од пациентите имаат и бришење на гените и трансформација makrogennye. Класичната форма solteryayuschaya 21-хидроксилаза дефицит е поврзан со точкести мутации, бришења или ген конверзија, што доведе до остро намалување или отсуство на 21-хидроксилаза активност. Повеќето пациенти со дефицит на 21-хидроксилаза се kompaudnymi хетерозиготна (м. Е. Тие имаат висока загуба на гени во било која од алели R450s21V). Постојат фенотипската варијабилност манифестации - solteryayuschaya virilizing форма, едноставна форма virilnoe вирилизација или одложени, што е манифестација на степенот на ензимски дефицит. На вториот вид се одредува кај пациенти со хетерозиготна мутации во kompaudnymi помалку функционално значајни алели R450s21V. генот недостаток R450s21 (21-хидроксилаза) не само што е тесно поврзан со HLA комплекс, но кај пациенти со 21-хидроксилаза дефицит беше откриено значително зголемување на содржината на некои специфични ХЛА подтипови. Тие вклучуваат Bw51 со едноставна форма на мажественост, Bw47 на solteryayuschey форма и Б14 со некласични форми VDKN.
Хидроксилаза дефицит R450s21 со вирилизација и губење на електролити
Solteryayuschaya формираат R450s21 дефицит хидроксилаза е откриен во околу 80% од пациентите со класичен дефицит на 21-хидроксилаза забележано со значајна активност R450s21 хидроксилаза дефицит доведува до оштетување на лачењето на кортизол и dosterona al. Веќе на 5-ти ден од животот што доведува до губење на електролити и течности и, според тоа, хипонатремија, хиперкалемија, ацидоза, дехидрација и циркулаторен колапс. Во ретки случаи, таа се развива подоцна, на 6-12 недели старост, кои обично се поврзани со придружно физиолошки стрес. Високата содржина на фетусот на андрогени до 12 недела од бременоста доведува до вирилизација на клиторисот и малите усни се спојуваат фетусот женски, по можност преку делумна употреба на "задна врата" за синтеза DHT е опишано Auchus и soavt.- андрогинот изложеност по 12-та недела на бременоста е само хипертрофија на клиторисот. Маскулинизација на надворешните гениталии кај жени со болеста е потешка отколку кај пациенти со едноставни или не solteryayuschey форма на неуспех R450s21 хидроксилаза. Кај мажите со оваа форма VDKN означени хипертрофија на пенисот. Често кај пациенти со дефицит на 21-хидроксилаза и доживотно учење-хидроксилаза означени хипофункција на кората на надбубрежните жлезди поради ниско vnutrinadpochech прекари концентрации на кортизол и дефекти развој и формирање на кора.
Неодамнешните студии покажаа дека содржината на надбубрежната жлезда феталниот намалена ензим SP-хидроксистероид дехидрогеназа-2 (3 (3-GSD2), кој води до секреција на DHEA-S, кој пак aromatize плацентата за да се произведе естроген. Активност на плацентарна вкус-PS се зголемува со бременоста, заштита на фетусот од ефектите на андрогени. Според најновите Оди et al., нормално женскиот фетус е заштитена со лачењето на адреналните андрогени во раните фази од бременоста (8-10 недели) минливи секреција LA-GSD2 надбубрежните жлезди, кога ниту една zkaya активност плацентата ароматаза уште се ниски, и диференцијација надворешниот фетусот гениталиите веќе се случува. Ова води кон минливи лачење на кортизол и базалните инхибиција на секрецијата на ACTH со планираната истовремена намалување на лачењето на DHEA-S страна на надбубрежните жлезди на фетусот. По 10 недели од бременоста израз SP-GSD2 фетална надбубрежна намалена и зголемена секреција на DHEA-S и плацентарна ароматаза активност. Сепак биосинтетско нарушувања во кортизол синтеза на неуспехот на 21-хидроксилаза, 11-хидроксилаза, или почесто опишани недостаток на Р450 оксидоредуктаза повлекува прекумерно лачење андроген во текот на критичниот период (8-12 недели-I), кога плацентарна ароматаза активност и понатаму на ниско ниво.
недостаток хидроксилаза R450s21 со вирилизација
Едноставна форма virilnoe R450s21 инсуфициенција (21-хидроксилаза) манифестира синтеза намалување на кортизол, што претставува зголемување во содржината и да се зголеми АЦТХ секреција на андрогени и прекурсори од тоа. Инциденцата е 1: 50.000, што е околу 20% од лицата со "класични" форма R450s21 неуспех. Обично девојките со оваа болест е наведено помалку сериозни маскулинизација на надворешните гениталии во споредба со solteryayuschey форма R450s21 неуспех. Кај машките фетуси на раѓање не го означи повреда на структурата на надворешните гениталии, но последиците од можно хипертрофија на фалусот. Овие пациенти обично се синтетизираат доволно за да се спречи симптомите неуспех минералокортикоидно износ алдостерон, иако тие може да имаат некоја дефект синтезата neralokortikoidov Е, кој служи како маркер на покачени нивоа на ренин во крвната плазма. Кај пациенти кои не се на третман, по раѓањето на вирилизација продолжува. Ова доведува до брзо зголемување на коскената возраст, и појавата на симптомите на вишокот на секрецијата андрогените (како што се акни, себореа, прекумерна развој на мускулното ткиво dopubertatnogo срамни и аксиларните раст на косата и хипертрофија на пенисот). Децата со оваа форма VDKN со peripubertatnym коскената возраст по почетокот на glucocorticoid терапија може да започне точно (централно) предвремен сексуален развој.
Некласичните недостаток хидроксилаза R450s21
Постојат случаи на делумен неуспех R450s21 (21-хидроксилаза). Можеби како симптоматска (одложен или не-класична) или асимптоматски ( "латентна" форма) на болеста. Овие "меки" форми на неуспех R450s21 хидроксилаза поврзани со HLA, како и на "класичен" форма на неуспех R450s21 gidroksilazy- Сепак, оваа варијанта на болеста е почеста отколку класичните. Познато е дека на недостаток "некласичните" форми R450s21 хидроксилаза е најчестиот автосомно рецесивно заболување. Се верува дека зачестеноста на појавата на некласични форми R450s21 хидроксилаза дефицит е 1 во 27 во Ашкенази Евреите, од 1 до 53 во шпански, 1 333 Италијанци и 1 во 1000 и во другите европски земји. Фреквенцијата на хетерозиготи некласичните форми можат да стигнат до 1:60. Пациенти со nonclassic R450s21 хидроксилаза дефицит често unexpressed хомозиготни за мутација (на пример, V281L) или се kompaudnymi хетерозиготи со другите позначајни скриени мутирани алели (27-76%). Жените со одложено манифестации на неуспех R450s21 хидроксилаза прослави раѓањето на нормалната структура на надворешните гениталии и не електролитен дисбаланс. Слабо изразена вирилизација започнува подоцна во детството или адолесценцијата, и доведува до предвремено појавата срамни и аксиларните дистрибуција на косата, умерена хипертрофија клиторисот, менструални нарушувања, акни, хирзутизам, полицистични јајници и да се забрза коска возраст. На не-класична форма на неуспехот R450s21 хидроксилаза е прогресивна болест. Мажите со оваа болест имаат нормална машки гениталии на раѓање, расте брзо, и тие прослави прерано созревање на скелетот. Подоцна почнува предвремена раст на косата на срамни и пазувите, предвремен пубертет со несразмерно мали тестиси и прекумерна развојот на мускулното ткиво. Иако во детството што го забележа релативно висок раст, додека растат имаат тенденција да имаат низок раст се должи на напредувањето на возраста на коските и предвремено затворање на раст зони. Кога хормонални или генотипизација студии на семејства во кои се појавува болеста, ги идентификува лицата со асимптоматска, но со биохемиски нарушувања кои се слични на блага форма на R450s21gidroksilazy неуспех.
} {Модул direkt4
недостаток на дијагноза хидроксилаза R450s21 секогаш треба да биде во следниве случаи: (1) од пациентите со двојна гениталии со кариотип 46, XX (на пример, пациенти со 46, XX НДП ..) - (2) момчиња со очигледни kriptorhizmom- (3) Деца пропадне државата со хипогликемија и биохемиски абнормалности карактеристика на надбубрежните nedostatochnosti- (4) момчиња и девојки пред пубертет virilizing симптоми, вклучувајќи предвремени adrenar тој. Рана дијагноза на неуспех R450s21 хидроксилаза израснат во идентификување на висока содржина на 17-ketosteroids и pregnantriola во урината. Иако дефиницијата на содржината на стероиди во урината се уште е во побарувачката и е потребно, овој метод беше заменет од страна на повеќе едноставни, евтини и ефикасни дефинирање на содржината во крвната плазма 17-хидроксипрогестерон, андростенедион и тестостерон.
На концентрација на 17-хидроксипрогестерон се зголеми во крв од папочна врвца, но се намалува брзо и да достигне вредности од 100-200 ng / dl (3.6 mmol / L) за 24 часа по раѓање. Децата на породување и новороденчиња во стрес нивоа на 17-хидроксипрогестерон се обично повисоки во споредба со децата е стресна. Пациенти со R450s21 хидроксилаза количина недостаток на 17-хидроксипрогестерон од 5.000 ng / dL (150 mmol / l) и зависи од возраста на пациентот и степенот на инсуфициенција R450s21 хидроксилаза. Пациенти со недостаток R450s21 хидроксилаза (форми на крајот на почетокот или асимптоматски) може да имаат гранична базалните вредности на 17-хидроксипрогестерон, сепак, како што е демонстрирано во Њу et al., Тие можат да се разликуваат од хетерозиготна најголемиот емисијата на 17-хидроксипрогестерон одговор на парентерална администрација ACTH.
Solteryayuschie форми може да се дијагностицира врз основа на клинички симптоми или хипонатремија и хиперкалемија во биохемиска анализа на крвта кај деца со нормална исхрана. Во одговор на концентрацијата на натриум ниски серумски во такви лица се намали на алдостерон во крвта и урината, и плазма ренин активност е значително зголемен. Во многу мајчиното млеко и детето формула обично ниска содржина на натриум.
HLA пишување, одредување на 17-хидроксипрогестерон во амнионската течност, и хорионски ресички биопсија со HLA-tipiruyut vaniem и генетски анализи употребени во пренатална дијагноза со фетална лезии. Клиничките студии покажале дека во раните фази на бременост, пренатална дексаметазон терапијата, доби мајка, може да го намали степенот на оштетување на формирање на гениталиите забележани кај новородени девојки со zabolevaniem- сепак користење на овој вид на третман останува контроверзно. Друштвото на детска ендокринологија и Lawson Вилкинс Европското здружение за педијатриски ендокринолози се согласија дека, и покрај очигледната непосреден ефект намали маскулинизација на женски надворешни гениталии на фетусот кога мајката доби дексаметазон, потребни се лонгитудиналните студии со цел да се избегне долгорочни несакани ефекти.
Хетерозиготна може да се идентификува со користење на ХЛА типизација во семејства со историја оптоварени, ACTH-индуцирана содржина пораст од 17-хидроксипрогестерон 21-деоксикортизол и крвна плазма, како и генетските анализи. Зголемување на серумските концентрации на 21-деоксикортизол повеќе информативна отколку 17-хидроксипрогестерон, и е во моментов на располагање во комерцијални лаборатории. Одредување на нивото на 17-хидроксипрогестерон во суви капиларна крв место на филтер-хартија со помош на специјален тест ленти е сигурен метод за скрининг на 21-хидроксилаза дефицит кај новороденото. Меѓутоа, кога скрининг поради стрес-индуцирана и постнатална нивоа обновување на 17-хидроксипрогестерон можноста за лажно-позитивни резултати. Кога скрининг обично не се откриени кај пациенти со не-класичните форми на 21-хидроксилаза дефицит, како и некои пациенти со класична форма, кој го прослави подоцна зголемување од 17-хидроксипрогестерон.
Хидроксилаза дефицит R450c11
Класичната форма на неуспех R450s11 хидроксилаза (вирилизација и хипертензија) е ретка и е 1: 100.000 деца во Европа. Сепак, во Централна Азија, болеста е многу почеста. Кај пациенти со класична форма на дефект болест 11-хидроксилаза резултира со намалување на концентрациите на кортизол, под влијание на зголемување на содржината на ACTH хиперсекреција и 11 dezoksikor tikosterona-и 11-деоксикортизол, и надбубрежните андрогени. Опишан значителна клиничка хетерогеност и хормонални манифестации, вклучувајќи класична, доцна, па дури и асимптоматски форми. Пациентите со оваа форма на адренална хиперплазија обично се подложни на вирилизација (поради хиперпродукција на андрогени) и хипертензија поврзана со зголемување на лачењето на 11-деоксикортикостеронот. Плазма ренин обично е нормална или намалена. Хипертензија може да се изрази во различни степени, беше забележано во две од трите случаи, и може да се поврзани со hypokalemic алкалоза.
R450s11 два гени се наоѓа на долгиот крак на хромозомот 8: R450s11b1 и R450s11b2. Како 21-хидроксилаза ген, два гени се 95% хомологна. P450s11b1 кодира ензимот 11-хидроксилација на зракот и се изразува во гломеруларна области е ACTH-зависни. Првично тоа посредува 11-Gi-droksilirovanie 11-деоксикортизол (со формирање на кортизол) и деоксикортикостерон (кортикостерон за да се формира). Таа има околу 1/12 P450c11b2 можности во врска со 18-Gi-droksilirovaniya и оксидира во 18-gidroksikor tikosteron на алдостерон. P450c11b2 го кодира на ангиотензин-зависни изоензим алдостерон синтаза и се изразува само во гломеруларната зона, каде што го потенцира 11-хидроксилација на 18-хидроксилација и 18-оксидација. Мутации, бришења и дуплирање на овие гени може да доведе до различни клинички манифестации на virizatsii и хипертензија (инсуфициенција P450c11b2) за да се изолиран соли загуба (инсуфициенција P450c11b2 - aldosteronsynthetase) или гликокортикоиди потисната хипертензија (со поврзување на ACTH-зависни регулаторни ген дел 11-хидроксилаза од кодирани дел aldosteronsynthetase). Двата гени кои кодираат P450c11b1 и P450c11b2, кој се наоѓа на хромозомот 8 и затоа не се поврзани со HLA. Примерокот со стимулација на ACTH не открива постојан биохемиски абнормалности докажаа хетерозиготи.
Дијагноза неуспех P450c11b1-хидроксилаза е потврден со зголемување на базалните или ACTH-индуцирана концентрација на 11-деоксикортизол и 11-деоксикортикостеронот, во крвната плазма на не помалку од 3 пати во споредба со 95 перцентил на релевантната возрасна група и зголемување на екскрецијата на метаболити во урината (главно , тетрахидро-11-деоксикортизол).
Хидроксистероид дехидрогеназа H
Машки или женски псевдохермафродитизам и адренална инсуфициенција дискутира подолу.
Недостаток на 17,20-лиаза P450c17
Машки псевдохермафродитизам, сексуална инфантилност, хипертензија и hypokalemic алкалоза се дискутира подолу.
Недостаток на ѕвезда и P450scc
Поврзани масното адренална хиперплазија, машки псевдохермафродитизам, сексуална инфантилизам и адренална инсуфициенција се дискутира подолу.
Недостаток на Р450 оксидоредуктаза
POR е флавопротеин која служи за пренос на електрони од никотинамид аденин динуклеотид фосфат (NADP) на сите микрозомални цитохроми P450. Во 1985 година, Peterson et al. Ние се опише дете со двојна гениталии, која, очигледно, имаше истовремена недостаток на 17- и 21-хидроксилаза. Милер вели дека тоа дете имаше дефект, а кофактор на двете ензими, наместо мутации во гените. Fluck, Милер и сор. 4 прв пат е опишана, а подоцна и во комбинација со 32 поединечни неуспех 17- и 21-хидроксилаза поради POR мутации. Сите пациенти кои се утврди содржината на сексуални стероиди, документирани покачени серумски концентрации на прогестерон и 17-хидроксипрогестерон во позадина на нормална или намалена содржина на С19 стероидите во крвта, со што се потврдува отсуството на 17- и 21-хидроксилаза. Имаше генотипските женски вирилизација, додека генотипските мажите - несоодветни маскулинизација.
Многумина, но не сите пациенти имаат дизморфизам: хипоплазија на черепот на лицето, кранијални и lokteluchevoy synostosis. Фенотип наречена "синдром Antle-Bixler" со скелетни дизморфизам. Дисморфична синдром Antle-Bixler, веројатно предизвикана од страна на две различни генетски дефекти. Лица со нормална гениталии имаат мутација на фибробластниот фактор на раст од 2, додека пациентите со синдромот Antle-Bixler и двојна гениталии имаат мутации во POR и треба да се гледа од страна на ендокринолог, како тие имаат зголемен ризик од адренална инсуфициенција, особено во услови на стрес. Мајките на децата со променети POR се изложени на ризик на вирилизација за време на бременоста, кои често е поврзан со дефицит на ароматаза (POR-зависни ензими).
Гледано во женски новороденчиња со овој дефект не е прогресивен вирилизација по раѓањето, за разлика од другите virilizuyu-ИНГ форми VDKN. Homma et al., Анализа на содржината на стероиди во урината на 5 пациенти, открил постоењето на синтеза "бајпас" пат дихидротестостерон кај пациенти со недостаток на Р450 оксидоредуктаза. Дефиниран опсег на стероиди во плазмата уште ви овозможува да се дијагностицира оваа болест. Повеќето пациенти се дијагностицира со маса спектроскопија стероиди во урината следи Генетски потврда. Спектарот на фенотипски манифестации продолжува да се прошири, како што некои пациенти имаат благи или без заболувања на коските се нормални гениталии и експлицитна сексуална дисфункција во зрелоста. А широк спектар на формирање генитални аномалии често зависи од степенот на дисфункција R450s21, P450c17 и П450 ароматаза, која зависи од сериозноста на промени во генот на Р450 оксидоредуктаза.
третман
Третман на пациенти со дисфункција на кората на надбубрежните во акутни и хронични фази на болеста варира. Во акутна адренална мозочен удар кортизол и алдостерон недостаток предизвикува хипогликемија, хипонатремија, хиперкалемија, хиповолемија, шок и ацидоза. Ако пациентот има хипогликемија треба да влезат во гликоза интравенски 0,25-0,5 g / kg (максимум 25 g). Ако пациентот е во состојба на колапс, можно е во првиот час интравенска инфузија на солена вода (20 ml / kg) - тогаш се компензира недостатокот на глукоза, електролити и течности од недостасува базирани бази и стандардните барања. Хидрокортизон натриум сукцинат, 50 mg / m2 треба да се даде болус, а потоа како што следува 50-100 mg / m2 за 24 часа е додадена на терапија на инфузија. Во присуство на хипонатремија и хиперкалемија може да се користи внатре сам 0,05-0,1 mg флудрокортизон или во комбинација со интравенска хидрокортизон. Од хидрокортизон минералокортикоидно активност, таа може да ги надомести и точни нарушувања на електролитите кога се администрира со солена вода. Во акутни случаи, хипонатремија, хиперкалемија и ацидоза може да има потреба администрација од натриум бикарбонат и размена на катјонска смола (м. E. Polisterinovogo натриум сулфонат). Пациенти со претходните хронична хипонатремија треба да биде внимателно и полека се зголеми на дофат на својата содржина, бидејќи на можноста за mielolizisa понс. Првично, пациенти со ново дијагностицирана болест и кај новороденчиња во текот на првите денови од третманот обично се користи хидрокортизон во дози од 50 mg / m2 / ден за да ја спречи активноста на оската хипоталамо-хипофизата-надбубрежните.
По постигнување на стабилизација и дијагноза врз основа на утврдување на стероиди, пациентот треба да примаат дози на одржување на глукокортикоидите за нормален раст, развој и вработување на коскената маса (хидрокортизон, околу 10-15 mg / m2 / ден орално во 3 дози). Дозата на хидрокортизон се пресметува врз основа на содржината на стероидните хормони во плазмата и урината, годишната добивка во раст, збир на коскената маса и клинички симптоми со стероиди или virilizing предозирање. Кога сол со губење на embodiments е потребен третман рудар-lokortikoidami (флудрокортизон 0,05-0,2 mg / ден орално) и опционална формирање на сол со храна (1-3 g / ден) за време на детството. минералокортикоидно доза треба да се прилагоди за да се постигне нормални вредности Содржина на електролити и крвниот притисок, како и активноста на ренин во крвната плазма. Неодамна, како резултат на комплексноста на "оптимална" третман на овие пациенти, беа предложени нови тераписки режими. Овие вклучуваат адреналектомија кај пациенти со мутации во R450s21, со користење на Somatotropin да се зголеми завршно раст, употребата на физиолошки дози на хидрокортизон (8 mg / m2), флудрокортизон, флутамид, и комбинации на андрогениот рецептор блокатори и инхибитори на ароматаза.
Во пост-пубертетски пациенти со гликокортикоиди терапија за замена може да се врши повеќе активни препарати кои имаат подолг полу-живот во споредба со хидрокортизон - како што се преднизон или метилпреднизолон, тие може да се користи 2 пати на ден. Како и со деца, треба најниска дози на дрога, доволни за оптимална супресија на надбубрежната секреција андрогени и спречување на несакани ефекти на стероиди. Дозата се пресметува во зависност стероиди во плазма, или уште подобро, со содржина од 17-ketosteroids во урината - индикатор за 24-часовна секреција на андрогени. Како и во случајот на хидрокортизон, дозата за секој пациент ќе зависи од апсорпција, метаболизам и чувствителност на дрога, и варира од пациент до пациент. Родителите треба да се обезбеди "целосна слика" на дијагноза, медицинска историја, можни терапевтски и хируршки третман, ризици, компликации и непознати моменти. Тоа е од витално значење дека тие имаат информации и време за да се донесе одлука за секоја операција нивното дете.
Само за пациенти со очигледни двојна надворешните гениталии (Prader III-V) ја разгледува можноста за хируршки третман. Прикажани vaginoplasty и клиторичен рецесија или klitoroplastika но не klitorektomiya! Сепак, може да се повтори хируршка корекција на гениталиите по интервенција произведени дете. При извршување на хируршки третман е да се воспостави функција, не само за да се елиминира козметички дефект. Операцијата треба да се врши од страна на искусен хирург кој ја разбира важноста на одржување на функционален интегритет на гениталиите. Лојд et al. неодамна го опиша различни "нормална" големина и изглед на надворешните гениталии од жените. Оваа студија може да се формира на основа на индикациите за операција кај возрасни пациенти со VDKN. Изборот на хируршки пат е контрадикторен. Оваа одлука треба да биде донесена по разговорите со родителите тактичен. Таа е опишана, но не е докажано, дека "козметички" хирургија намалува родителска вознемиреност и подобрување на нивниот однос со детето. Тоа е важно да се разговара со своите родители, постојат лекарите и група на адвокати кои се многу препорачува да не се врши никаква хируршка интервенција (ако тоа не е "медицински потребни") се додека детето е доволно стари за да направите свој одлука . Треба да се напомене дека оваа препорака непознат долгорочни резултати. Главен приоритет на семејството со болно дете треба да бидете сигурни дека растот и развојот на детето ќе доведе до формирање на возрасен со оптимална функционалност.
Истражување Беренбаум et al., Dessens et al., Маер-Bahlburg et al. и Хајнс покажаа дека огромното мнозинство од 46, XX жени со 21-хидроксилаза дефицит и сродни вирилизација на надворешните гениталии имаат женски родов идентитет. Кај овие пациенти, иако ретко, но почесто отколку во општата популација, ги исполнува тендер дисфорија. Однесувањето маскулинизација / defeminizatsiya повеќе типични форми solteryayuschih VDKN споредба со nesolteryayuschimi форми.
Со соодветен третман на пациенти со најчестиот облик на VDKN - 21 gidrokslazy дефицит - можете да очекувате нормален плодноста кај мажите и нормално феминизација, менструални функција и плодноста кај жените. Во споредба со општата популација на пациенти со плодноста VDKN малку намален се должи на неколку фактори, не најмалку на која е намалувањето на мајките инстинкт. Ние треба долгорочна психолошка помош и поддршка на пациентот и на членовите на неговото семејство.
Во машките пациенти со неуспех R450s21 хидроксилаза под стимулирачки ефект на ACTH може да го зголеми остаток ткиво од тестисите надбубрежните локализација кои може да се помешаат со тестикуларен тумор. Надбубрежните ткиво обично се наоѓа на билатерално ниво и се состои од клетки кои не се разликуваат хистолошки од Лајдиговите клетки, освен отсуство на кристалоиди Reinke. Обично ова се случува не се платени или не дисциплинирани пациенти. За да се спречи оваа компликација, како и спречување на адренална криза, Bazo хипофизата-надбубрежните philous хиперплазија и карцином, машки пациенти во зрелоста препорачува континуирана терапија со гликокортикоиди (и, доколку е индицирано, минералокортикоидно).
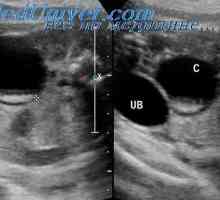 Крипторхизам кај фетусот. тестикуларна феминизација
Крипторхизам кај фетусот. тестикуларна феминизација Конгенитална адренална хиперплазија. Лечење на адренална хиперплазија.
Конгенитална адренална хиперплазија. Лечење на адренална хиперплазија. Virilizing конгенитална адренална хиперплазија: Причини и механизми за развој
Virilizing конгенитална адренална хиперплазија: Причини и механизми за развој Диференцијална дијагноза и третман на virilizing адренална хиперплазија
Диференцијална дијагноза и третман на virilizing адренална хиперплазија Адренална хиперплазија клиника virilizing и неговите форми
Адренална хиперплазија клиника virilizing и неговите форми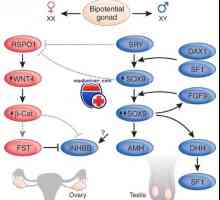 Мутација и ген дуплирање dax1, sox9. пол генотип несовпаѓање xy и kampomelicheskaya дисплазија
Мутација и ген дуплирање dax1, sox9. пол генотип несовпаѓање xy и kampomelicheskaya дисплазија Недостаток на 11-бета-хидроксилаза - еден од причини на конгенитална адренална хиперплазија (CAH)
Недостаток на 11-бета-хидроксилаза - еден од причини на конгенитална адренална хиперплазија (CAH)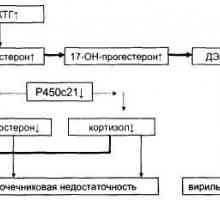 Особено конгенитална адренална хиперплазија
Особено конгенитална адренална хиперплазија- Adrenogenital синдром (конгенитална адренална хиперплазија, конгенитална адренална хиперплазија)…
- Здравствени енциклопедија, болест, лекови, лекар, аптека, инфекција, извадоци, секс, гинекологија,…
- Здравствени енциклопедија, болест, лекови, лекар, аптека, инфекција, извадоци, секс, гинекологија,…
- Здравствени енциклопедија, болест, лекови, лекар, аптека, инфекција, извадоци, секс, гинекологија,…
- Здравствени енциклопедија, болест, лекови, лекар, аптека, инфекција, извадоци, секс, гинекологија,…
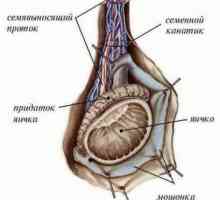 Диференцијацијата на тестисите и јајниците
Диференцијацијата на тестисите и јајниците Хирзутизам и virilization
Хирзутизам и virilization Надбубрежните вирилизација
Надбубрежните вирилизација Конгенитална адренална хиперплазија: симптоми, третман, дијагноза
Конгенитална адренална хиперплазија: симптоми, третман, дијагноза Конгенитална адренална хиперплазија кај возрасни: симптоми, третманот, причини, симптомите
Конгенитална адренална хиперплазија кај возрасни: симптоми, третманот, причини, симптомите Кариотип 46 xx жена
Кариотип 46 xx жена Leydig клеточна аплазија
Leydig клеточна аплазија Конгенитални малформации на гениталните органи
Конгенитални малформации на гениталните органи
 Особено конгенитална адренална хиперплазија
Особено конгенитална адренална хиперплазија Хетеросексуални сексуалниот развој
Хетеросексуални сексуалниот развој Хирзутизам и virilization
Хирзутизам и virilization Конгенитална адренална хиперплазија. Лечење на адренална хиперплазија.
Конгенитална адренална хиперплазија. Лечење на адренална хиперплазија.