Жолтица кај дете со Тирозинемија
наследни Тирозинемија
Cодржина
сите новороденчиња од областа на Saguenay и во 28% од пациентите спојување мутација е идентификуван во другите региони на светот. Повеќето од овие пациенти се хомозиготни за оваа мутација, гванин да аденин беше заменет во нуклеотидна секвенца на спојување донатор. Според работата на други истражувачи, човечки fah ген има должина од 35 kb и е поделена на 14 егзони. Означени нуклеотидна мутации со замена на тимидин да гванин и триптофан на стоп кодон на позиција 262.
изразување fah во црниот дроб на пациентите со наследна Тирозинемија беше анализирана во неколку молекуларна нивоа, вклучувајќи mRNA и ензимската активност, што резултира во диференцијација на фенотипски варијанти. Деца со Тирозинемија Опишани се многу генетски мутации FAH- fah израз хетерогеност во црниот дроб на пациенти регистрирани на ниво на mRNA, протеини и ензимската активност.
До неодамна, немаше методи на лекување на децата со наследна Тирозинемија, освен дестинација почетокот на диета ограничување на тирозин и трансплантација на црниот дроб. Клонирање на cDNA кодирање fah трансфер генот користење ретровирус интеграза, и го направи можно за иднината методи на генетска терапија за оваа болест.
Веќе, можете да го направите на активност fah фибробластите на пациенти со недостаток на овој ензим.
На прв документиран метод на терапија наследни Тирозинемија Тоа беше употребата на дериватот на пестициди (NTBC, или nitizinona [2- (2-нитро-4-trifluorometilbenzol) -1,3-tsiklogeksanedion]). Оваа хемиско соединение е казнив со инхибиција на метаболизмот на тирозин-4 gidroksifenilpiruvatdioksigenazy инхибирање на појава и акумулација suktsinilatsetona и suktsinilatsetoatsetata. Во првата клиничка студија, пациентите добиле nitizinon орално во доза од 0,1-0,6 mg / kg / ден.
серум Suktsinilatseton брзо се намали на речиси невидливи нивоа, покажале подобрување на клиничката состојба во целина, и функцијата на црниот дроб, како и намалено ниво на серумските АФП. По првичниот успех на третманот на наследни Тирозинемија nitizinonom во Гетеборг (Шведска) беше спроведено мултицентрична студија вклучува речиси 300 пациенти од целиот свет. Многу од овие пациенти биле третирани за 5 години со добри резултати. Таа во моментов е познато колку нови фармаколошки лекови ќе влијае на способноста за развој на HCC.

како HCC хистолошки потврдена кај 37% од децата со Тирозинемија, резултатите од студијата Гетеборг укажуваат на значително пониска инциденца на болеста кај оние пациенти кои nitizinon доделен на 2-годишна возраст. моментално nitizinona Препорачана доза е 1 mg / kg / ден. Лекот се користи против диета со рестрикција на фенилаланин и тирозин.
Видео: Неонатална жолтица. билирубин броеви. © Наталија Shilov
трансплантација на црн дроб врши деца со Тирозинемија во случај на акутна хепатална инсуфициенција, слаб одговор на фармакотерапијата и наводната HCC. Трансплантација на црн дроб се решава проблемот за тоа како се препорачува тумор на црниот дроб, и метаболички нарушувања наследни Тирозинемија тип I. Пред терапевтска употреба nitizinona трансплантација на црн дроб за сите деца со наследен Тирозинемија на возраст од 12 месеци.
И покрај значителниот подобрување на контрола на метаболизмот благодарение nitizinonom терапија на HCC уште претставува сериозен ризик за сите пациенти со наследна Тирозинемија. Nitizinon не корегира абнормални генската експресија и хепатална дисплазија. HCC ризик се зголеми за одложување на почетокот на терапијата nitizinonom, па затоа е неопходно да се назначат штом тоа станува возможно.
Класично наследни Тирозинемија Таа има хронични и акутни форми. Пациенти со хронична ниво имунореактивните fah е околу 20% од нормалното ензимската активност. Пациентите со акутна форма на болеста не е откриен на immunoblots имунореактивните fah црниот дроб, бубрезите и лимфоцити. Пред широката употреба nitizinona со наследна Тирозинемија шанси за преживување на пациентите само во диететика тоа беше прилично ниско.
преживување по изглед симптомите варира во зависност од возраста на која се појави болеста, - претходно се појавуваат симптомите, на полошо прогноза. Најчести причини за смрт се црниот дроб и истовремена крварење (67%), HCC (17%) и слични порфирија со синдром на респираторна недостаток (10%). стапка од една година преживување по појавата на болеста кај деца на возраст од 2 месеци, помеѓу 2 и 6 месеци, а повеќе од 6 месеци беше 38, 74 и 96%, соодветно. Тирозинемија форми кои се појавуваат многу рано, почетокот и крајот, врз основа на овие услови за опстанок на новата класификација беше предложен.
Видео: Продолжениот жолтица. Мајчиното млеко жолтица. © Наталија Shilov
по добивањето во јануари 2002 година, одобрение од американската ФДА да се применуваат на забележаните nitizinon реално подобрување на морбидитетот и морталитетот поради наследни Тирозинемија.
На акутна форма на наследна Тирозинемија Тоа може да се случи на било која возраст. Обично тоа води до тешка хепатоцелуларна дисфункција и често се поврзува со значително зголемување на серумскиот ниво АФП. тирозин нивоа во серумот се исто така најчесто се покачени. Дијагностичка карактеристика во овој вродени грешки на метаболизмот е значително зголемување на содржината на тирозин suktsinilatsetona во урината.
На 48 деца со 42% е забележано акутен невролошки нарушувања (Криза), не е опишано претходно како компликација. Оваа состојба се развива во просечна возраст од 1 година и ги има следниве симптоми: периферна невропатија, придружени со болка и повраќање, и хипертензија razgibateley- ileus- парализираниот мускулна слабост и обиди за самоповредување. Помеѓу кризи, повеќето деца останаа непроменети.
сигурен биохемиски маркер на невролошки кризи Тоа не е пронајден, иако се испитува нивото на тирозин и suktsinilatsetona во урината. Уринарна екскреција на 8-aminolevulinic киселина (невротоксични средно во биосинтезата на порфиринот) е подигната за време на кризи, но истиот се зголеми, исто така, е забележано кај асимптоматски периоди. Се смета дека suktsinilatseton можат да имаат невротоксични својства. Интересно, студии на животни откриваат фактот дека мозокот е конфискуван suktsinilatsetonom и дека повредата на биосинтезата на темата во мозокот може да доведе до влошување на оксидативниот метаболизам. Така, suktsinilatseton може да биде одговорен за симптомите на ЦНС Тирозинемија.
Во хронична форма на наследна Тирозинемија Може да се идентификуваат на гломеруларна и тубуларна дисфункција, и вклучување во патолошки процес црниот дроб, нервниот и мускулниот систем. Кај деца со долгорочни дисфункција на бубрезите може да се развие тешка интерстицијална болест. Во ера кога nitizinona рано именување на лекот кај повеќето пациенти задржа нормална функција на бубрезите тубули и неколку деца треба трансплантација на бубрег.
 Абнормалности на чорапи гени и синдром на TVH Holt-Орам. Фактори Фибробластен раст
Абнормалности на чорапи гени и синдром на TVH Holt-Орам. Фактори Фибробластен раст Tanatofornaya дисплазија на фетусот. Причини и инциденцата на дисплазија tanatoformnoy
Tanatofornaya дисплазија на фетусот. Причини и инциденцата на дисплазија tanatoformnoy 10% На вродени срцеви заболувања не се наследени од родителите
10% На вродени срцеви заболувања не се наследени од родителите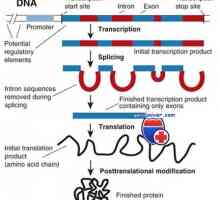 Транскрипција. Форми и видови на РНК клетки
Транскрипција. Форми и видови на РНК клетки Формирање на два синџири на ДНК. генетскиот код
Формирање на два синџири на ДНК. генетскиот код Миграција повреди на ентеричен нервен систем во болеста на Хиршпрунг
Миграција повреди на ентеричен нервен систем во болеста на Хиршпрунг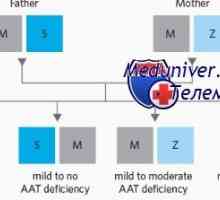 Жолтица дете во недостаток на алфа-1-антитрипсин (А1-часот)
Жолтица дете во недостаток на алфа-1-антитрипсин (А1-часот) Неонатална жолтица со метаболички нарушувања
Неонатална жолтица со метаболички нарушувања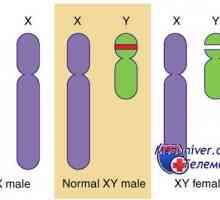 Генетски нарушувања на гонадите. Гените SRY, WT1 и синдроми Фрејзер и Denis-dresha
Генетски нарушувања на гонадите. Гените SRY, WT1 и синдроми Фрејзер и Denis-dresha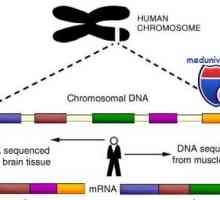 Гени и човечки хромозом. структура
Гени и човечки хромозом. структура Мутации на гонадотропин гени. Мутации во подединици на LH и FSH
Мутации на гонадотропин гени. Мутации во подединици на LH и FSH Недостаток на миелопероксидаза (МПО). Клиника и дијагностика
Недостаток на миелопероксидаза (МПО). Клиника и дијагностика- Ефект на мутации во RET-ефикасноста на терапијата со медуларен рак на тироидната жлезда
 Главните пристапи за пренатална дијагноза на генетски болести. Директен дијагноза на генетски…
Главните пристапи за пренатална дијагноза на генетски болести. Директен дијагноза на генетски…- Gaucher-ова болест се однесува на болести sfingolipidozam lipidov- акумулацијата поради дефект на…
- Меркаптопурин (mercaptopurinum). 6-меркаптопурин (монохидрат). Синоними: leykerin, leukerin,…
- Здравствени енциклопедија, болест, лекови, лекар, аптека, инфекција, извадоци, секс, гинекологија,…
- Onkologiya-
- Onkologiya-
 Повреди на амино киселина метаболизам
Повреди на амино киселина метаболизам Мутации кои водат до наследни болести кај луѓето
Мутации кои водат до наследни болести кај луѓето
 Формирање на два синџири на ДНК. генетскиот код
Формирање на два синџири на ДНК. генетскиот код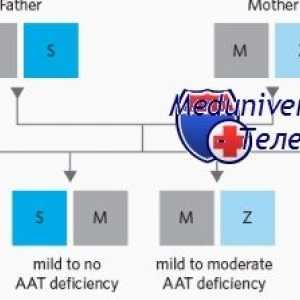 Жолтица дете во недостаток на алфа-1-антитрипсин (А1-часот)
Жолтица дете во недостаток на алфа-1-антитрипсин (А1-часот) Главните пристапи за пренатална дијагноза на генетски болести. Директен дијагноза на генетски…
Главните пристапи за пренатална дијагноза на генетски болести. Директен дијагноза на генетски… Мутации кои водат до наследни болести кај луѓето
Мутации кои водат до наследни болести кај луѓето Неонатална жолтица со метаболички нарушувања
Неонатална жолтица со метаболички нарушувања Абнормалности на чорапи гени и синдром на TVH Holt-Орам. Фактори Фибробластен раст
Абнормалности на чорапи гени и синдром на TVH Holt-Орам. Фактори Фибробластен раст Tanatofornaya дисплазија на фетусот. Причини и инциденцата на дисплазија tanatoformnoy
Tanatofornaya дисплазија на фетусот. Причини и инциденцата на дисплазија tanatoformnoy