Amavroticheskaya вродени идиотизам. муколипидоза
Neyrovistseralny GM3-ганглиозидоза, ензимски дефект е непозната. Се карактеризира со продолжена разбира, прогресивно намалување во визија, спленомегалија и лимфаденопатија. Хистолошки преглед на внатрешните органи покаже "пенлив" клетки, како клетките Ниман - Трансферот. Сепак, за разлика од болест на Ниман - Изберете клетки во одложено ганглиозиди GM3 и GM2 и sfipgomielin не одложен присутен макуларна цреша-црвена дамка.
Amavroticheskaya вродени идиотизмот - ганглиозидоза GD3 (disialodigeksozid ганглиозидоза). Тоа се случува многу ретко. дефект ензим не е разбрана. Биохемиските природата на депозити во ткивата не е јасно. Симптомите на болеста се констатирани веднаш по раѓањето (аритмија дишење, цијаноза, недостаток на цицање рефлекс и дисфагија). Гледано конвулзии. Цреша-црвена дамка во фундусот е отсутен, има атрофија на оптичкиот нерв.
смрт Тоа се случува во првите неколку месеци од животот. Грубо: атрофија на церебрална хемисфера и малиот мозок со хидроцефалус образование во областа разредена кора цистична кариес. Хистолошки незрели (фетопатија) кората на мозокот клетки потечени состојат чување производи. Постојат индикации за присуство на Судан-годишно различни специјализирани "пенлив" клетки во слезината, црниот дроб, белите дробови, тимусот, бубрезите и надбубрежните жлезди, што укажува на генерализирана процес.
на мозочните неврони PAS-позитивни, sudanofilnoe супстанција. Кога биохемиска анализа откри абнормални мозокот ганглиозидни GD3.
муколипидоза --GM1 ганглиозидоза. Во оваа форма не само се акумулира ганглиозидоза GM1 ганглиозидите, мукополисахариди, туку првенствено кератан сулфат, urinable, GM1 ганглиозидоза тоа зафаќаат средна позиција меѓу ганглиозидоза и мукополисахаридоза [Gafhmann Х. et al., 1976]. На ензим има дефект во системот A, B и C фракции галактозидаза. Овие податоци служат како повод за да се потенцира во 1970 година, независна тип tezaurismoza наречен муколипидоза. Муколипидоза во кршење на GM1 ганглиозидни размена на познатите во форма на два вида на одредени ензимски дефекти.

Генерализирана-GM1 ганглиозидоза Тип I со синдром psevdogurlera. Во 1964 година тој го опиша Landig наречен семејство neyrovistseralnogo липидоза. Во 1965 година O`Brien покажа дека ова е ганглиозидоза акумулирано ганглиозидни GM1. Ензимски дефект лежи во целосно отсуство на фракции A, B и C галактозидаза. Болеста почнува со раѓањето, брзо напредува оштетување на мозокот, што резултира со смрт во првите 2 години од животот. Има промени во скелетот, слични на синдромот Хурлер (груби црти на лицето, хипертрофија на долната вилица, лумбална кифосколиоза, хипертрофија на епифизеална 'рскавица).
Покрај тоа, постојат gepato- спленомегалија. Хистолошки во ганглиските клетки утврдени шик-позитивни и sudapofilny материјал со последователна смрт на невроните и развојот на мозокот на глиоза.
Во црниот дроб, слезината, тимусот, лимфните јазли, коскената срцевина, бубрежна епител тубули најде "пенлив" клетки sudanofilnye. Електрон-микроскопски преглед за откривање на невроните и олигодендроцитите osmiophil теле со ламеларна и зрнести компоненти проширување цитоплазматски ретикулум и перинуклеарно простори врши оптички густа тела. Биохемиски можност да се покаже дека износот на GM1 ганглиозидни во мозокот се зголемува 20-30 пати во однос на норма, зголемување на содржината GM2-ганглиозид и кератан сулфат.
Малолетничка форма GM1 ганглиозидоза-тип 2. дефект на ензимот е уред од фракции B и C бета-галактозидаза. Болеста почнува со крајот на првата година од животот со симптоми на атаксија, прогресивна деменција, губење на видот и губење на моторна активност. Смртта се јавува во 3-8 години. Скелетните промени се утврдува само во текот на радиографија ( "клунови" дното лумбален 'рбетниот тела). Хепато-и спленомегалија не се случи. Хистолошки, за разлика од другите оштетувањето на невроните преовладува ганглиозидоза малиот мозок следи глиоза. Во мозокот содржина GM1 ганглиозидни е зголемен за 10 пати во споредба со норма.
 Ганглиозидоза. Sandhofa болест и малолетничка ганглиозидоза
Ганглиозидоза. Sandhofa болест и малолетничка ганглиозидоза Муколипидоза: mannozidoz и фукозидоза. Малолетничка sulfatidoz тип oustina
Муколипидоза: mannozidoz и фукозидоза. Малолетничка sulfatidoz тип oustina Дијагноза sulfatidoza. Sfingomielinoz Ниман-Пикова болест
Дијагноза sulfatidoza. Sfingomielinoz Ниман-Пикова болест И leukodystrophies мукополисахаридоза. Ризик од мукополисахаридоза
И leukodystrophies мукополисахаридоза. Ризик од мукополисахаридоза Http://sweli.ru/media/k2/items/cache/977bf03741e8c7acdb0cd307a5e8d8d0_s.jpgизменения плацентата со…
Http://sweli.ru/media/k2/items/cache/977bf03741e8c7acdb0cd307a5e8d8d0_s.jpgизменения плацентата со… Карактеристики на абнормалности на оптичкиот нерв во развојот
Карактеристики на абнормалности на оптичкиот нерв во развојот- Акутна циркулаторниот нарушувања во крвните садови на ретината
- Атрофија на оптичкиот нерв. Етиологија. Заболувања на оптичкиот нерв и ретината, болест на мозокот,…
- Ден-слепило (ноќно слепило, ноќно слепило) -rasstroystvo самракот визија. Етиологијата на вродени…
- Конгестивна оптичкиот диск nerva- noninflammatory papilledema. Етиологија: болести на централниот…
- Опструкција на централната артерија setchatkizakuporka централната ретинална артерија поради…
- Дива цреша плодови (fructus Ради). Собрани во текот на целосна созревање и сушено овошје од диви и…
- Здравствени енциклопедија, болест, лекови, лекар, аптека, инфекција, извадоци, секс, гинекологија,…
- Здравствени енциклопедија, болест, лекови, лекар, аптека, инфекција, извадоци, секс, гинекологија,…
- Здравствени енциклопедија, болест, лекови, лекар, аптека, инфекција, извадоци, секс, гинекологија,…
 Повреда на метаболизмот на липидите
Повреда на метаболизмот на липидите Ниман-Пиковата болест
Ниман-Пиковата болест Neyrometabolicheskie болест
Neyrometabolicheskie болест Оштетувањето на оптичкиот нерв, симптомите
Оштетувањето на оптичкиот нерв, симптомите Вродени макуларна промени
Вродени макуларна промени Муколипидоза
Муколипидоза
 Оштетувањето на оптичкиот нерв, симптомите
Оштетувањето на оптичкиот нерв, симптомите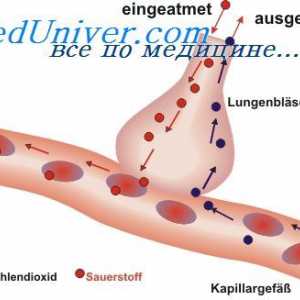 Дијагноза sulfatidoza. Sfingomielinoz Ниман-Пикова болест
Дијагноза sulfatidoza. Sfingomielinoz Ниман-Пикова болест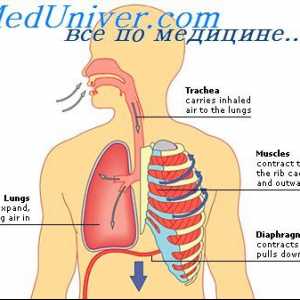 Муколипидоза: mannozidoz и фукозидоза. Малолетничка sulfatidoz тип oustina
Муколипидоза: mannozidoz и фукозидоза. Малолетничка sulfatidoz тип oustina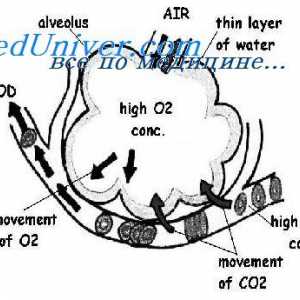 И leukodystrophies мукополисахаридоза. Ризик од мукополисахаридоза
И leukodystrophies мукополисахаридоза. Ризик од мукополисахаридоза Вродени макуларна промени
Вродени макуларна промени Neyrometabolicheskie болест
Neyrometabolicheskie болест